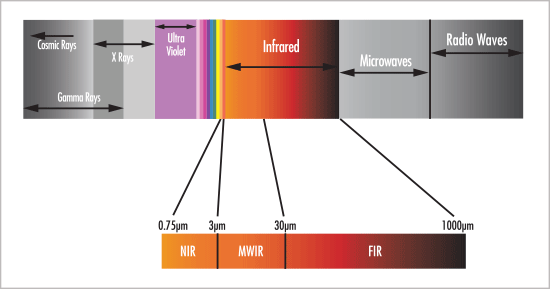
Another blog not about NIR? Near-infrared (NIR) spectroscopy is a secondary method. That means that NIR doesn't measure things like fat, protein, moisture, ash, %-polymerization or anything else directly. Instead, we use an acceptable primary method to train our NIR to make those measurements. More details on NIR calibration can be found in this earlier... Continue Reading →
Primary Method Feature: Extraction (Part 1: Extraction Foundations)